Simple RNA-Seq pipeline
Last updated on 2024-12-04 | Edit this page
Estimated time: 60 minutes
Overview
Questions
- How can I create a Nextflow pipeline from a series of unix commands and input data?
- How do I log my pipelines parameters?
- How can I manage my pipeline software requirements?
- How do I know when my pipeline has finished?
- How do I see how much resources my pipeline has used?
Objectives
- Create a simple RNA-Seq pipeline.
- Use the
log.infofunction to print all the pipeline parameters. - Print a confirmation message when the pipeline completes.
- Use a conda
environment.ymlfile to install the pipeline’s software requirement. - Produce an execution report and generate run metrics from a pipeline run.
We’re now set to develop a multi-step pipeline using Nextflow, for analyzing and performing quality control on our yeast RNA-Seq experiment.
In this RNA-Seq pipeline, we’ll undertake the following steps to thoroughly analyze gene expression data:
- Quality Control with FastQC: FastQC assesses the quality of the data by generating reports that highlight any potential issues, such as low-quality sequences or contamination. FastQC’s output includes an HTML report and a directory containing detailed analyses, which are essential for evaluating the integrity of the sequencing data.
- Transcriptome Indexing with Salmon: Salmon is a tool to quantify transcript expression in RNA-seq data. The first step in the process is for Salmon to create an index of the transcriptome. This step involves processing a reference transcriptome, which allows for efficient and accurate mapping and quantification of RNA-Seq reads.
- Quantification with Salmon: After indexing, Salmon is used for the quantification step. In this step, Salmon maps the reads to the transcriptome index and quantifies the transcript abundances. This process is crucial for determining the expression levels of genes in the sample.
- Aggregating Reports with MultiQC: Finally, the pipeline employs MultiQC to aggregate logs and outputs from the FastQC and Salmon. MultiQC scans the outputs and compiles a summary report, which provides an overview of the results and highlights any areas that may need further investigation.
This pipeline provides an overview of RNA-Seq data, from quality control to expression quantification, culminating in an aggregated report for easy interpretation of the results.
To start move the episode’s nextflow scripts in the
scripts/rnaseq_pipeline folder to your home directory.
This folder contains files we will be modifying in this episode.
Define the pipeline parameters
The first thing we want to do when writing a pipeline is define the
pipeline parameters. The script script1.nf defines the
pipeline input parameters.
GROOVY
//script1.nf
params.reads = "data/yeast/reads/*_{1,2}.fq.gz"
params.transcriptome = "data/yeast/transcriptome/*.fa.gz"
println "reads: $params.reads"Run it by using the following command:
We can specify a different input parameter using the
--<params> option, for example :
OUTPUT
reads: data/yeast/reads/ref1*_{1,2}.fq.gzAdd a parameter
Modify the script1.nf adding a third parameter named
outdir and set it to results. This parameter
will be used as the pipeline output directory.
It can be useful to print the pipeline parameters to the screen. This
can be done using the the log.info command and a multiline
string statement. The string method .stripIndent() command
is used to remove the indentation on multi-line strings.
log.info also saves the output to the log execution file
.nextflow.log.
log.info
Modify the script1.nf to print all the pipeline
parameters by using a single log.info command and a
multiline string statement. See an example here.
Look at the output log .nextflow.log.
Create a transcriptome index file
Nextflow allows the execution of any command or user script by using
a process definition.
For example,
A process is defined by providing three main declarations:
The second example, script2.nf adds,
- The process
INDEXwhich generate a directory with the index of the transcriptome. This process takes one input, a transcriptome file, and emits one output a salmon index directory. - A queue Channel
transcriptome_chtaking the transcriptome file defined in params variableparams.transcriptome. - Finally the script adds a
workflowdefinition block which calls theINDEXprocess.
GROOVY
//script2.nf
/*
* pipeline input parameters
*/
params.reads = "data/yeast/reads/*_{1,2}.fq.gz"
params.transcriptome = "data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz"
params.outdir = "results"
println """\
R N A S E Q - N F P I P E L I N E
===================================
transcriptome: ${params.transcriptome}
reads : ${params.reads}
outdir : ${params.outdir}
"""
.stripIndent()
/*
* define the `INDEX` process that create a binary index
* given the transcriptome file
*/
process INDEX {
input:
path transcriptome
output:
path 'index'
script:
"""
salmon index --threads $task.cpus -t $transcriptome -i index
"""
}
transcriptome_ch = channel.fromPath(params.transcriptome)
workflow {
INDEX()
}Try to run it by using the command:
OUTPUT
N E X T F L O W ~ version 22.04.0
Launching `script2.nf` [happy_brown] DSL2 - revision: 90e932bb8d
R N A S E Q - N F P I P E L I N E
===================================
transcriptome: data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz
reads : data/yeast/reads/*_{1,2}.fq.gz
outdir : results
Process `INDEX` declares 1 input channel but 0 were specified
-- Check script 'script2.nf' at line: 41 or see '.nextflow.log' file for more detailsThe execution will fail because the program the process,
INDEX , has not been passed any input channel.
Add the transcriptome_ch channel to the
INDEX process call.
Now try to run it again by using the command:
Now the workflow will run successfully.
OUTPUT
N E X T F L O W ~ version 22.04.0
Launching `script2.nf` [mad_aryabhata] DSL2 - revision: 811396b67b
R N A S E Q - N F P I P E L I N E
===================================
transcriptome:data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz
reads : data/yeast/reads/*_{1,2}.fq.gz
outdir : results
executor > local (1)
[c0/418d78] process > INDEX (1) [100%] 1 of 1 ✔
The INDEX process also defines one output
channel. This channel will be populated with index
directory created during process. To view the contents of the channel we
can use the view operator.
View the contents of the index_ch
- Assign the output of the
INDEXprocess to the variableindex_ch. - View the contents of the
index_chchannel by using theviewoperator.
Collect read files by pairs
This step shows how to match read files into pairs, so they can be mapped by salmon.
The script script3.nf adds a line to create a channel,
read_pairs_ch, containing fastq read pair files using the
fromFilePairs channel factory.
GROOVY
//script3.nf
nextflow.enable.dsl = 2
/*
* pipeline input parameters
*/
params.reads = "data/yeast/reads/ref1_{1,2}.fq.gz"
params.transcriptome = "data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz"
params.outdir = "results"
log.info """\
R N A S E Q - N F P I P E L I N E
===================================
transcriptome: ${params.transcriptome}
reads : ${params.reads}
outdir : ${params.outdir}
"""
.stripIndent()
read_pairs_ch = Channel.fromFilePairs( params.reads )We can view the contents of the read_pairs_ch by adding
the following statement as the last line:
Now if we execute it with the following command:
It will print an output similar to the one shown below that shows how
the read_pairs_ch channel emits a tuple. The tuple is
composed of two elements, where the first is the pattern matched by the
glob pattern data/yeast/reads/ref1_{1,2}.fq.g, defined by
the variable params.reads , and the second is a list
representing the actual files.
OUTPUT
[..truncated..]
[ref1, [data/yeast/reads/ref1_1.fq.gz,data/yeast/reads/ref1_2.fq.gz]]To read in other read pairs we can specify a different glob pattern
in the params.reads variable by using --reads
options on the command line. For example, the following command would
read in add the ref samples:
OUTPUT
[..truncated..]
[ref2, [data/yeast/reads/ref2_1.fq.gz, data/yeast/reads/ref2_2.fq.gz]]
[ref3, [data/yeast/reads/ref3_1.fq.gz, data/yeast/reads/ref3_2.fq.gz]]
[ref1, [data/yeast/reads/ref1_1.fq.gz, data/yeast/reads/ref1_2.fq.gz]]Note File paths including one or more wildcards ie.
*, ?, etc. MUST be wrapped in single-quoted
characters to avoid Bash expanding the glob pattern on the command
line.
We can also add a argument, checkIfExists: true , to the
fromFilePairs channel factory to return an message if the
file doesn’t exist.
GROOVY
//script3.nf
[..truncated..]
read_pairs_ch = Channel.fromFilePairs( params.reads, checkIfExists: true )If we now run the script with the --reads parameter
data/yeast/reads/*_1,2}.fq.gz
it will return the message .
OUTPUT
[..truncated..]
No such file: data/yeast/reads/*_1,2}.fq.gzRead in all read pairs
- Add the
checkIfExists: trueargument to thefromFilePairschannel factory inscript3.nf. - Using the command line parameter
--reads, add a glob pattern to read in all the read pairs files from thedata/yeast/readsdirectory.
OUTPUT
[..truncated..]
[temp33_1, [data/yeast/reads/temp33_1_1.fq.gz, data/yeast/reads/temp33_1_2.fq.gz]]
[ref2, [data/yeast/reads/ref2_1.fq.gz, data/yeast/reads/ref2_2.fq.gz]]
[temp33_3, [data/yeast/reads/temp33_3_1.fq.gz, data/yeast/reads/temp33_3_2.fq.gz]]
[ref3, [data/yeast/reads/ref3_1.fq.gz, data/yeast/reads/ref3_2.fq.gz]]
[temp33_2, [data/yeast/reads/temp33_2_1.fq.gz,data/yeast/reads/temp33_2_2.fq.gz]]
[etoh60_2, [data/yeast/reads/etoh60_2_1.fq.gz,data/yeast/reads/etoh60_2_2.fq.gz]]
[ref1, [data/yeast/reads/ref1_1.fq.gz, data/yeast/reads/ref1_2.fq.gz]]
[etoh60_3, [data/yeast/reads/etoh60_3_1.fq.gz, data/yeast/reads/etoh60_3_2.fq.gz]]
[etoh60_1, [data/yeast/reads/etoh60_1_1.fq.gz, data/yeast/reads/etoh60_1_2.fq.gz]]Perform expression quantification
The script script4.nf;
- Adds the quantification process,
QUANT. - Calls the
QUANTprocess in the workflow block.
GROOVY
//script4.nf
..truncated..
/*
* Run Salmon to perform the quantification of expression using
* the index and the matched read files
*/
process QUANT {
input:
each index
tuple val(pair_id), path(reads)
output:
path(pair_id)
script:
"""
salmon quant --threads $task.cpus --libType=U -i $index -1 ${reads[0]} -2 ${reads[1]} -o $pair_id
"""
}
..truncated..
workflow {
read_pairs_ch = Channel.fromFilePairs( params.reads, checkIfExists:true )
transcriptome_ch = Channel.fromPath( params.transcriptome, checkIfExists:true )
index_ch=INDEX(transcriptome_ch)
quant_ch=QUANT(index_ch,read_pairs_ch)
}The index_ch channel, declared as output in the
INDEX process, is used as the first input argument to the
QUANT process.
The second input argument of the QUANT process, the
read_pairs_ch channel, is a tuple composed of two elements:
the pair_id and the reads.
Execute it by using the following command:
You will see the execution of the index and quantification process.
Re run the command using the -resume option
The -resume option cause the execution of any step that
has been already processed to be skipped.
Try to execute it with more read files as shown below:
OUTPUT
N E X T F L O W ~ version 21.04.0
Launching `script4.nf` [shrivelled_brenner] - revision: c21df6839e
R N A S E Q - N F P I P E L I N E
===================================
transcriptome: data/yeast/transcriptome/Saccharomyces_c
erevisiae.R64-1-1.cdna.all.fa.gz
reads : data/yeast/reads/ref*_{1,2}.fq.gz
outdir : results
executor > local (8)
[02/3742cf] process > INDEX [100%] 1 of 1, cached: 1 ✔
[9a/be3483] process > QUANT (9) [100%] 3 of 3, cached: 1 ✔You will notice that the INDEX step and one of the
QUANT steps has been cached, and the quantification process
is executed more than one time.
When your input channel contains multiple data items Nextflow, where possible, parallelises the execution of your pipeline.
In these situations it is useful to add a tag directive
to add some descriptive text to instance of the process being run.
Add a tag directive
Add a tag directive to the QUANT process of
script4.nf to provide a more readable execution log.
Data produced by the workflow during a process will be saved in the
working directory, by default a directory named work. The
working directory should be considered a temporary storage space and any
data you wish to save at the end of the workflow should be specified in
the process output with the final storage location defined in the
publishDir directive.
Note: by default the publishDir
directive creates a symbolic link to the files in the working this
behaviour can be changed using the mode parameter.
Add a publishDir directive
Add a publishDir directive to the quantification process
of script4.nf to store the process results into folder
specified by the params.outdir Nextflow variable. Include
the publishDir mode option to copy the
output.
Challenge
Recap
In this step you have learned:
How to connect two processes by using the channel declarations.
How to resume the script execution skipping already already computed steps.
How to use the
tagdirective to provide a more readable execution output.How to use the
publishDirto store a process results in a path of your choice.
Quality control
This step implements a quality control step for your input reads. The
input to the FASTQC process is the same
read_pairs_ch that is provided as input to the
quantification process QUANT .
GROOVY
//script5.nf
[..truncated..]
/*
* Run fastQC to check quality of reads files
*/
process FASTQC {
tag "FASTQC on $sample_id"
cpus 1
input:
tuple val(sample_id), path(reads)
output:
path("fastqc_${sample_id}_logs")
script:
"""
mkdir fastqc_${sample_id}_logs
fastqc -o fastqc_${sample_id}_logs -f fastq -q ${reads} -t ${task.cpus}
"""
}
[..truncated..]
workflow {
read_pairs_ch = Channel.fromFilePairs( params.reads, checkIfExists:true )
transcriptome_ch = Channel.fromPath( params.transcriptome, checkIfExists:true )
index_ch=INDEX(transcriptome_ch)
quant_ch=QUANT(index_ch,read_pairs_ch)
}Run the script script5.nf by using the following
command:
The FASTQC process will not run as the process has not
been declared in the workflow scope.
MultiQC report
This step collect the outputs from the quantification and fastqc steps to create a final report by using the MultiQC tool.
The input for the MULTIQC process requires all data in a
single channel element. Therefore, we will need to combine the
FASTQC and QUANT outputs using:
- The combining operator
mix: combines the items in the two channels into a single channel
GROOVY
//example of the mix operator
ch1 = Channel.of(1,2)
ch2 = Channel.of('a')
ch1.mix(ch2).view()OUTPUT
1
2
a- The transformation operator
collectcollects all the items in the new combined channel into a single item.
OUTPUT
[1, 2, 3]Combining operators
Which is the correct way to combined mix and
collect operators so that you have a single channel with
one List item?
quant_ch.mix(fastqc_ch).collect()quant_ch.collect(fastqc_ch).mix()fastqc_ch.mix(quant_ch).collect()fastqc_ch.collect(quant_ch).mix()
You need to use the mix operator first to combine the
channels followed by the collect operator to collect all
the items in a single item.
In script6.nf we use the statement
quant_ch.mix(fastqc_ch).collect() to combine and collect
the outputs of the QUANT and FASTQC process to
create the required input for the MULTIQC process.
GROOVY
[..truncated..]
//script6.nf
/*
* Create a report using multiQC for the quantification
* and fastqc processes
*/
process MULTIQC {
publishDir "${params.outdir}/multiqc", mode:'copy'
input:
path('*')
output:
path('multiqc_report.html')
script:
"""
multiqc .
"""
}
workflow {
read_pairs_ch = Channel.fromFilePairs( params.reads, checkIfExists:true )
transcriptome_ch = Channel.fromPath( params.transcriptome, checkIfExists:true )
index_ch=INDEX(transcriptome_ch)
quant_ch=QUANT(index_ch,read_pairs_ch)
fastqc_ch=FASTQC(read_pairs_ch)
MULTIQC(quant_ch.mix(fastqc_ch).collect())
}Execute the script with the following command:
OUTPUT
N E X T F L O W ~ version 21.04.0
Launching `script6.nf` [small_franklin] - revision: 9062818659
R N A S E Q - N F P I P E L I N E
===================================
transcriptome: data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz
reads : data/yeast/reads/*_{1,2}.fq.gz
outdir : results
executor > local (9)
[02/3742cf] process > INDEX [100%] 1 of 1, cached: 1 ✔
[9a/be3483] process > QUANT (quantification on etoh60_1) [100%] 9 of 9, cached: 9 ✔
[1f/b7b30a] process > FASTQC (FASTQC on etoh60_1) [100%] 9 of 9, cached: 1 ✔
[2c/206fef] process > MULTIQC [100%] 1 of 1 ✔It creates the final report in the results folder in the
${params.outdir}/multiqc directory.
Handle completion event
This step shows how to execute an action when the pipeline completes the execution.
Note: that Nextflow processes define the execution of asynchronous tasks i.e. they are not executed one after another as they are written in the pipeline script as it would happen in a common imperative programming language.
The script script7..nf uses the
workflow.onComplete event handler to print a confirmation
message when the script completes.
GROOVY
workflow.onComplete {
log.info ( workflow.success ? "\nDone! Open the following report in your browser --> $params.outdir/multiqc/multiqc_report.html\n" : "Oops .. something went wrong" )
}This code uses the ternary operator that is a shortcut expression that is equivalent to an if/else branch assigning some value to a variable.
If expression is true? "set value to a" : "else set value to b"Try to run it by using the following command:
OUTPUT
[..truncated..]
Done! Open the following report in your browser --> results/multiqc/multiqc_report.htmlMetrics and reports
Nextflow is able to produce multiple reports and charts providing several runtime metrics and execution information.
The
-with-reportoption enables the creation of the workflow execution report.The
-with-traceoption enables the create of a tab separated file containing runtime information for each executed task, including: submission time, start time, completion time, cpu and memory used..The
-with-timelineoption enables the creation of the workflow timeline report showing how processes where executed along time. This may be useful to identify most time consuming tasks and bottlenecks. See an example at this link.The
-with-dagoption enables to rendering of the workflow execution direct acyclic graph representation. Note: this feature requires the installation of Graphviz, an open source graph visualization software, in your system.
More information can be found here.
Metrics and reports
Run the script7.nf with the reporting options as shown below:
- Open the file
report.htmlwith a browser to see the report created with the above command. - Check the content of the file
trace.txtor viewtimeline.htmlto find the longest running process. - View the dag.png
The INDEX process should be the longest running process.
dag.png 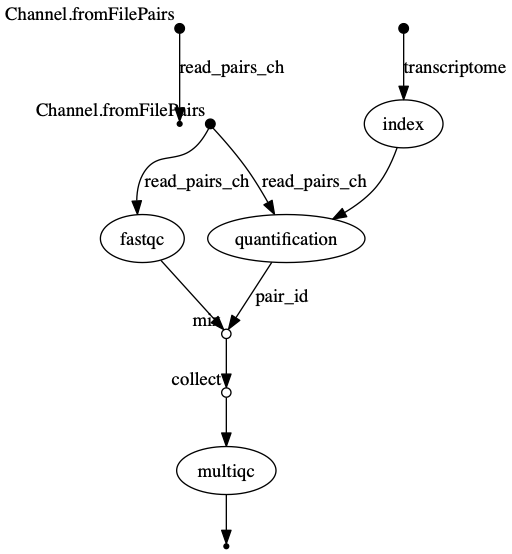 The vertices in the graph
represent the pipeline’s processes and operators, while the edges
represent the data connections (i.e. channels) between them.
The vertices in the graph
represent the pipeline’s processes and operators, while the edges
represent the data connections (i.e. channels) between them.
short running tasks
Note: runtime metrics may be incomplete for run short running tasks..
Key Points
- Nextflow can combined tasks (processes) and manage data flows using channels into a single pipeline/workflow.
- A Workflow can be parameterise using
params. These value of the parameters can be captured in a log file usinglog.info - Nextflow can handle a workflow’s software requirements using several
technologies including the
condapackage and enviroment manager. - Workflow steps are connected via their
inputsandoutputsusingChannels. - Intermediate pipeline results can be transformed using Channel
operatorssuch ascombine. - Nextflow can execute an action when the pipeline completes the
execution using the
workflow.onCompleteevent handler to print a confirmation message. - Nextflow is able to produce multiple reports and charts providing
several runtime metrics and execution information using the command line
options
-with-report,-with-trace,-with-timelineand produce a graph using-with-dag.